Síndrome hematofagocítico como manifestación inicial de linfoma de Hodgkin clásico
Hemophagocytic syndrome as the initial manifestation of Hodgkin lymphoma
DOI:
https://doi.org/10.11565/arsmed.v46i2.1744Palabras clave:
linfoma de Hodgkin, hematofagocitosis, virus de Epstein-BarrResumen
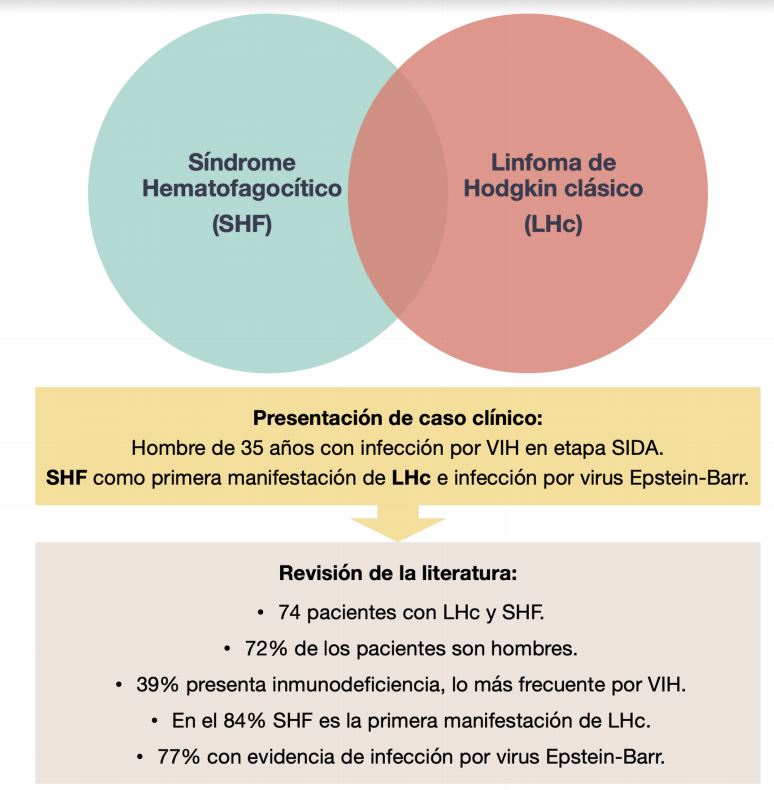
Introducción: las histiocitosis son un grupo heterogéneo de enfermedades; una de ellas es el síndrome hematofagocítico (SHF). Sus causas pueden ser infecciosas, neoplásicas, autoinmunes o relacionadas a inmunodeficiencias adquiridas; el linfoma de Hodgkin clásico (LHc) es una causa poco frecuente. Se reporta el caso de un hombre inmunodeprimido de 35 años que ingresa al hospital febril y con insuficiencia respiratoria grave.
Métodos: se recopiló información clínica pertinente y se revisó material de biopsia estudiado con tinción de hematoxilina – eosina, técnica inmunohistoquímica e hibridación in situ cromogénica. Resultados: estudios de laboratorio muestran pancitopenia, alteración de pruebas hepáticas, hipertrigliceridemia, hipoalbuminemia e hiperferritinemia. El estudio de médula ósea hematopoyética con mielograma y biopsia muestran hallazgos compatibles con LHc, signos de hemofagocitosis e infección por virus Epstein-Barr (VEB). Se diagnostica SHF como primera manifestación de LHc e infección por VEB. Conclusiones: a la fecha, se describen 74 pacientes reportados con SHF como manifestación de LHc; en el 84% fue su primera manifestación. Si bien la presentación clínica presentada es infrecuente, se ha propuesto una asociación en hombres con inmunodeficiencia, SHF, LHc e infección por VEB; por lo que se sugiere una alta sospecha diagnóstica.
Descargas
Descargas
Publicado
Cómo citar
Licencia
Derechos de autor 2020 ARS MEDICA Revista de Ciencias Médicas

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-SinDerivadas 4.0.
Los autores/as conservan sus derechos de autor y garantizan a la revista el derecho de primera publicación de su obra, la que estará simultáneamente sujeta a la Licencia CC BY-SA 4.0 (Ver declaración de Acceso Abierto).